
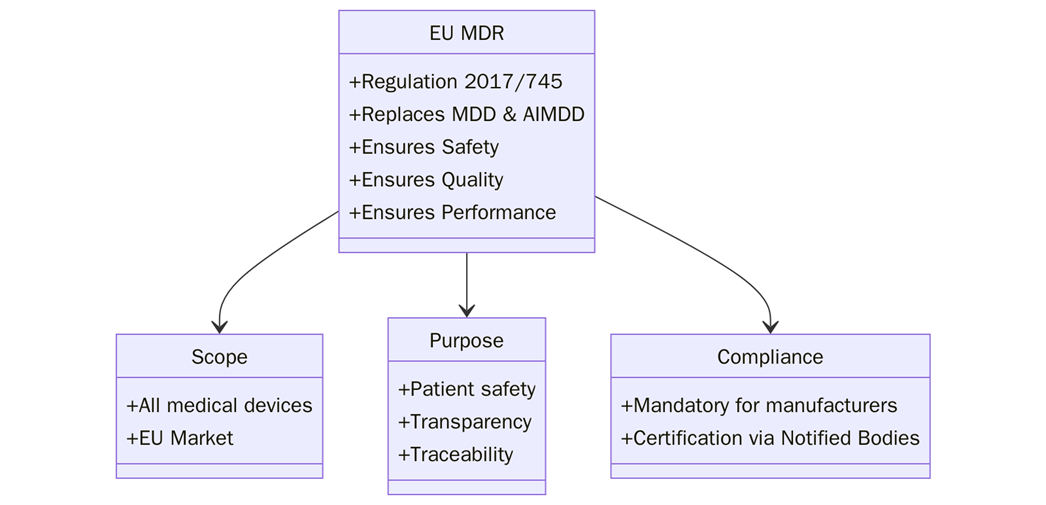
What is EU MDR?

The EU Medical Device Regulation (MDR), formally known as Regulation (EU) 2017/745, governs the medical device market in the European Union. The MDR sets out the requirements for the design, manufacture, and sale of medical devices within the EU. It ensures that manufacturers place devices on the market only after proving they meet high standards of safety and performance. The regulation also includes the requirements for conformity assessment, post-market surveillance, and vigilance.
One of the critical elements of the MDR is the technical file. It contains the information needed to prove that a medical device complies with regulatory requirements. Preparing and maintaining this file is vital for manufacturers who want to place medical devices on the European market.
For audits and certification, contact support@pacificcert.com.
Purpose of the technical File in EU MDR
The purpose of the technical file under the EU MDR is to provide a detailed record of the device’s design, manufacturing, performance, and safety data. It acts as proof that the device meets MDR requirements and complies with essential safety and performance rules.
The technical file also enables the Notified Body (NB) to assess the device for conformity. It verifies that the device complies with EU legislation before manufacturers can sell it. Besides being mandatory, a strong technical file supports regulatory authorities, market surveillance, and post-market activities.
Scope and Applicability
The EU MDR applies to all medical devices placed on the market within the European Union. It covers both low-risk products like Class I devices and high-risk products such as Class III devices. Manufacturers must prepare a technical file for every device regulated under the MDR. This file shows compliance with safety and performance requirements set by the regulation. The rule applies to EU manufacturers and to non-EU manufacturers. Non-EU companies must appoint an authorized representative in the EU to ensure the file is available and meets standards.
Key Definitions
- Technical File: A compilation of documents, design specifications, risk assessments, and other required data that show the safety and performance of a medical device.
- Essential Safety and Performance Requirements (ESRs): Manufacturers must meet these fundamental requirements before they can sell medical devices in the EU.
- Notified Body (NB): An independent, accredited organization authorized to conduct conformity assessments of higher-risk devices under the EU MDR.
- Conformity Assessment: The process of evaluating whether a device meets the requirements of the EU MDR, which may involve testing, inspections, and audits.
What are the requirements of Technical File Preparation under EU MDR?
To comply with the EU MDR, manufacturers must fulfill specific requirements while preparing the technical file. These requirements are essential to show the safety and performance of a medical device and ensure it meets regulatory standards. Below are the key requirements for preparing a overreaching technical file:
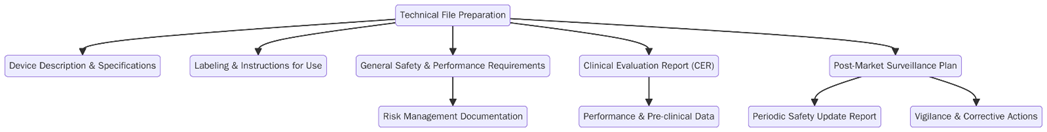
- The manufacturer must classify the device according to the MDR risk-based system (Class I, IIa, IIb, or III).
- The manufacturer must document and make traceable all aspects of the device’s design, manufacturing, testing, and distribution, keeping detailed records for each stage of the device’s lifecycle.
- The manufacturer must conduct a comprehensive risk assessment in line with ISO 14971 to identify potential hazards and record the measures taken to mitigate those risks.
- The manufacturer must include clinical data in the technical file to demonstrate the device’s safety and efficacy, following the MDR clinical evaluation guidelines.
- The manufacturer must establish a QMS that complies with MDR requirements and ensures consistent production of devices in accordance with regulatory standards.
- The manufacturer must set up a post-market surveillance system that explains how the device will be monitored for safety and performance once it enters the market.
- For higher-risk devices, the manufacturer must submit the technical file to a Notified Body for review and assessment before obtaining CE marking.
What are the benefits of Having a Overreaching Technical File?
A overreaching technical file ensures that the device is compliant with EU regulations, which helps maintain safety and quality standards. The benefits of preparing and maintaining a technical file include:
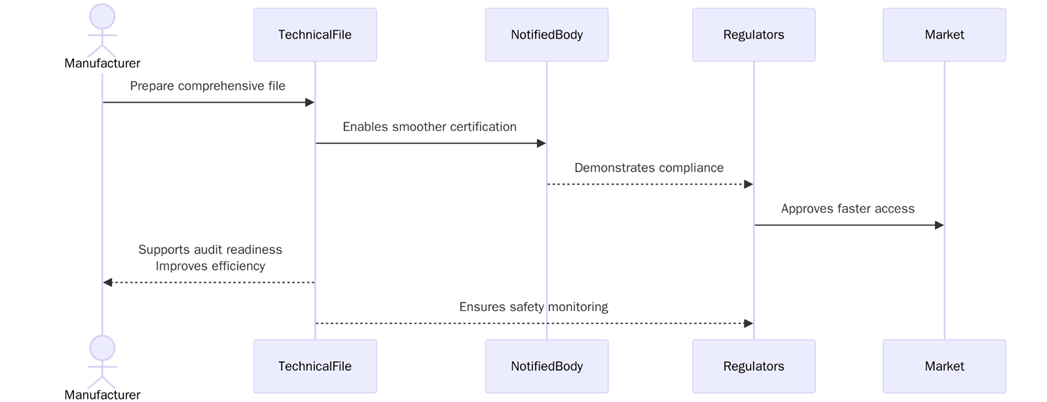
- A complete and compliant technical file is required to meet EU MDR regulations and gain market access in the European Union.
- A well-prepared technical file facilitates the conformity assessment process and can help avoid delays in obtaining CE marking.
- Demonstrating compliance with the EU MDR helps build trust with customers, stakeholders, and regulators, ensuring a positive market reputation.
- A well-documented technical file helps identify potential risks early in the development process and ensures that proper risk mitigation strategies are in place.
- By maintaining an up-to-date technical file, manufacturers can monitor the device’s safety and performance in the market, addressing issues quickly and effectively.
Recent trends in supply chain and distribution management emphasize the growing role of technology and automation in ensuring compliance with MDR. Manufacturers are increasingly adopting digital tools to streamline the technical file preparation process, making it easier to manage and update compliance documentation. As regulations become more stringent worldwide, the demand for technical files under the EU MDR is expected to grow. With a greater focus on post-market surveillance and clinical data, manufacturers will need to maintain up-to-date technical files throughout the product lifecycle. As global standards align with the EU MDR, technical file preparation will become a critical component of the regulatory landscape.
Certification Process for Technical File Preparation
The certification process involves the following steps:
- Initial Assessment: Conduct a thorough review of the device’s design, manufacturing, and risk management practices to identify any gaps in compliance.
- Documentation Gathering: Collect all relevant technical documentation, including clinical evaluations, risk assessments, and manufacturing processes.
- Submission for Notified Body Review: Submit the technical file to a Notified Body for conformity assessment and approval.
- Certification Awarded: Upon successful review, the Notified Body will issue a certificate of compliance, allowing the manufacturer to affix the CE mark to the device.
- Ongoing Monitoring: Maintain and update the technical file throughout the device lifecycle, particularly as new regulatory requirements are introduced.
Timeline for technical File Preparation
The typical timeline for preparing the technical file under the EU MDR can vary based on the complexity of the device and the organization’s preparedness.
Document Gathering takes 1-2 months for assembling the necessary documentation and ensuring completeness. Risk Management takes 2-3 months for conducting thorough risk assessments and mitigating identified risks. Manufacturers usually submit documentation to the Notified Body within 1–2 months, allowing time for submission and the initial review by the NB. Final Certification depends on the Notified Body’s review and any additional information required, certification may take 3-6 months.
What is the cost of Technical File Preparation?
The cost of preparing a technical file under the EU MDR can vary based on the size of the organization, the complexity of the device, and whether the device is subject to Notified Body review. Costs typically include:
Consulting and Documentation Fees is the fees for expert services to assist in the preparation and review of the technical file. Notified Body Fees is for notified Body assessment, especially for higher-risk devices (Class II and III). Costs for any additional regulatory assessments or submissions required for compliance is Regulatory Review Fees
How Pacific Certifications Can Help?
At Pacific Certifications, we provide overreaching auditing and certification services for EU MDR compliance. Our team will guide you through the entire technical file preparation process, ensuring that your medical device meets all required standards. Our services include:
- Stage 1 and Stage 2 audits to evaluate your manufacturing processes and ensure compliance with EU MDR.
- Objective conformity assessments based on the EU MDR guidelines.
- Certification issuance upon successful completion of the process.
- Ongoing surveillance audits to ensure continued compliance with EU MDR.
For audits and certification, contact support@pacificcert.com.
ISO 9001 and EU MDR Training and Courses
Various training courses are available to help organizations comply with EU MDR, including:
- Lead Auditor Training – Equips professionals to conduct external third-party audits.
- Lead Implementer Training – For those responsible for planning and executing ISO 9001 implementation.
- Internal Auditor Training – Preparing internal auditors for certification audits
Pacific Certifications provides accredited training programs. If your organization is looking for EU MDR training, our team is equipped to help you. Contact us at support@pacificcert.com.
Frequently Asked Questions (FAQs)
How long does it take to get EU MDR certification?
The process typically takes 6–12 months, depending on the complexity of the device and the readiness of the technical file.
Is EU MDR certification mandatory for all medical devices?
Yes, all medical devices sold in the EU must comply with the EU MDR, and a valid technical file is required for certification.
What are the main benefits of EU MDR certification?
EU MDR certification ensures product safety, regulatory compliance, increased market access, and consumer trust.
Can I apply for EU MDR certification without a complete technical file?
No, a complete technical file must be prepared and submitted to a Notified Body for review and certification.
How often do I need to update the technical file?
The technical file must be updated regularly, especially if there are changes to the design, manufacturing process, or regulatory requirements.
Ready to get certified?
Contact Pacific Certifications to begin your certification journey today!
Suggested Certifications –
Read more: Pacific Blogs





