Your practical guide to accredited medical device certification, trusted by regulators, manufacturers, and healthcare supply chains worldwide.
When your products are used in patient care, there is no margin for error. ISO 13485 certification shows that your Quality Management System has been independently assessed against the standard built specifically for medical devices, giving regulators, distributors, and healthcare providers confidence that safety and compliance sit at the centre of how you operate.
ISO 13485 is the international standard for Quality Management Systems specific to the medical devices industry. Unlike general quality standards, it was written with one purpose in mind: ensuring that medical devices consistently meet customer needs and the regulatory requirements that apply to them at every stage, from design and development through to production, storage, distribution, installation, and servicing.
The current version, ISO 13485:2016, places significant weight on risk management throughout the product lifecycle, traceability, and the kind of documented evidence regulators expect to see. While it shares some structural similarities with ISO 9001, it is not built on the Annex SL framework, and it does not place the same emphasis on customer satisfaction or continual improvement in the same open-ended way. Instead, it is built around regulatory compliance and product safety, which makes sense given what is at stake.
ISO 13485 applies to any organisation involved in the medical device supply chain, regardless of size. This includes manufacturers, design houses, contract packagers, sterilisation providers, distributors, and even organisations supplying components or services to device manufacturers.
Certification is recognised by regulatory bodies and notified bodies in major markets, and for many manufacturers, it is a practical prerequisite for getting a device onto shelves at all.
Get in touch with Pacific Certifications today for a seamless, transparent path to accredited registration. Our global experts are available 24/7 to support your operational needs.
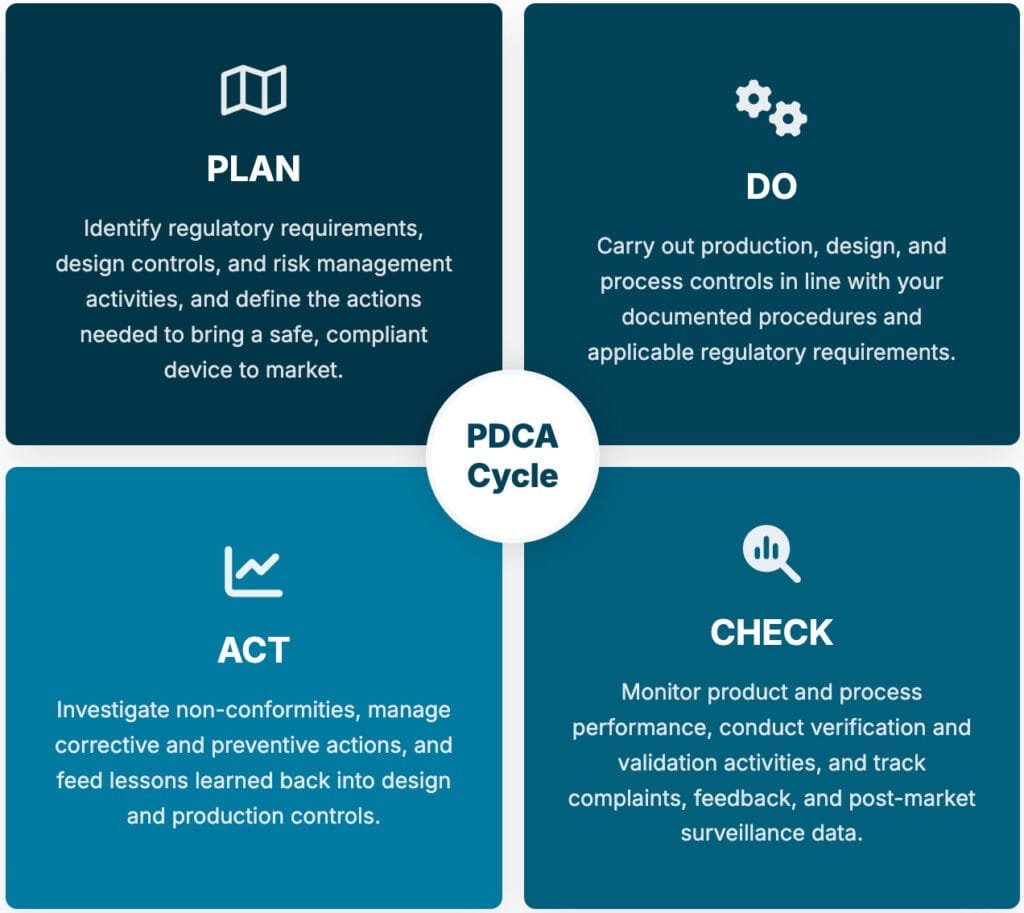
The PDCA (Plan-Do-Check-Act) cycle underpins ISO 13485 in much the same way it does other management system standards, but here it is applied with a sharper focus on patient safety and regulatory compliance at every turn.
From your first audit to ongoing surveillance, our auditors bring real medical device sector experience to every stage of your certification journey.
Receive an independently audited, internationally recognised ISO 13485 certificate through a clear, structured two-stage audit process.
Build internal expertise across your quality and regulatory teams with training pathways covering everything from awareness to lead auditor level.
Already certified to ISO 9001? We can align audit timing and reduce duplication where your systems overlap, without compromising on the rigour ISO 13485 demands.
Review your current QMS internally against ISO 13485 requirements before the formal audit, so design controls, risk files, and documentation are ready when it counts.
ISO 13485 has evolved to reflect tighter regulatory expectations and a sharper focus on risk across the full device lifecycle.
| Parameter | ISO 13485:2003 | ISO 13485:2016 |
|---|---|---|
| Risk Approach | Risk management referenced but loosely applied. | Risk-based thinking embedded across the entire QMS, not just product design. |
| Regulatory Alignment | General reference to applicable regulatory requirements. | Explicit requirements to identify and document applicable regulatory obligations for each market. |
| Outsourced Processes | Limited requirements for supplier and outsourced process control. | Stronger control and oversight requirements for suppliers and outsourced activities. |
| Software Validation | Minimal guidance on software used within the QMS. | Clear requirements for validating software used in the QMS, production, and monitoring. |
| Documentation | Standard documentation requirements similar to general QMS standards. | Expanded documentation, traceability, and record-keeping requirements throughout the device lifecycle. |
| Focus Area | General quality management adapted for medical devices. | Patient safety, regulatory compliance, and risk management as the central organising principles. |
ISO 13485 is shaped by a set of principles that prioritise patient safety and regulatory compliance above general business performance goals.
Risk must be considered and documented at every stage of the product lifecycle, from initial design through to post-market surveillance.
Organisations must identify and meet the specific regulatory requirements of every market they operate in, and keep pace as those requirements change.
Where outcomes cannot be fully verified by inspection alone, processes must be validated to confirm they consistently produce a safe, compliant result.
Devices, components, and materials must be traceable throughout the supply chain, enabling fast and accurate action if a safety issue arises.
Decisions, controls, and outcomes must be backed by documented evidence that regulators and notified bodies can review and rely on.
Organisations are responsible for the quality and compliance of outsourced processes and supplied components, not just their own internal operations.
Rather than open-ended improvement, the QMS must remain consistently suitable, adequate, and effective for its regulatory and patient safety purpose.
ISO 13485 follows an eight-clause structure, with Clauses 4 through 8 defining the requirements assessed during certification.
| Clause | Title | Scope & Requirement Objective |
|---|---|---|
| Clause 4 | Quality Management System | Establish, document, and maintain a QMS appropriate to the organisation's role in the medical device supply chain. |
| Clause 5 | Management Responsibility | Top management must demonstrate commitment, establish a quality policy, and ensure regulatory and customer requirements are met. |
| Clause 6 | Resource Management | Ensure adequate human resources, infrastructure, and work environment controls to support product safety and quality. |
| Clause 7 | Product Realization | Control planning, design and development, purchasing, production, and servicing of medical devices. |
| Clause 8 | Measurement, Analysis, and Improvement | Monitor product and process performance, manage complaints and non-conformities, and maintain QMS suitability through corrective action. |
The ISO 13485 requirements establish the essential controls for maintaining product quality, managing regulatory obligations and ensuring consistent performance throughout the medical device lifecycle.
Everything you need to prepare for ISO 13485 certification (pdf) in one place.
The path to certification balances system building with rigorous auditing.
Submit your application and tell us about your organisation, device classification, and applicable regulatory markets.
Before the formal audit begins, review your existing QMS internally against ISO 13485 requirements, including design controls and risk files, to identify and address any gaps.
A documentation review to confirm your QMS, regulatory file, and risk management documentation are adequately developed and ready for the on-site assessment.
Our auditor evaluates whether your QMS is fully implemented and effective across design, production, and post-market activities.
Upon successful completion, your ISO 13485 certificate is issued, valid for three years.
Annual surveillance audits maintain your certification, followed by full recertification at the end of the three-year cycle.
For a standard organisation, the certification process typically follows a ten-week timeline.
| Week | Activity | Core Milestones & Focus Areas |
|---|---|---|
| Week 1 | Application & Scoping | Submit your application and define the scope of your QMS, including applicable device classifications and markets. |
| Week 2 | Gap Analysis | Internally review existing QMS documentation, design controls, and risk files against ISO 13485 requirements. |
| Weeks 3-4 | QMS Implementation | Deploy or refine processes, complete risk management files, and ensure documented information is in place. |
| Weeks 5-6 | Stage 1 Audit | Our auditor reviews your QMS, regulatory file, and risk documentation to confirm readiness for the main assessment. |
| Weeks 7-8 | Stage 2 Audit | Full on-site or remote evaluation of your QMS implementation across design, production, and post-market processes. |
| Week 9 | Technical Review | Address any findings, close non-conformities, and finalise the certification review. |
| Week 10 | Certificate Issuance | Receive your accredited ISO 13485 certificate upon successful completion of the assessment. |
| Note: The timeline is indicative and may vary depending on device classification, organisational complexity, number of locations, documentation readiness, and completion of any corrective actions required. | ||
ISO 13485 certification cost varies depending on your organisation’s size, device classification, number of locations, and the complexity of your design and production processes. If you are integrating ISO 13485 with ISO 9001 or other standards under a single audit programme, this can also influence the overall cost.
At Pacific Certifications, we offer transparent, competitive pricing with no hidden charges. Contact us for a tailored quote or use our free cost calculator to get an instant estimate.
Regulatory scrutiny on medical devices continues to tighten across every major market. Manufacturers that cannot demonstrate a certified, risk-based Quality Management System face longer approval timelines, more difficult distributor relationships, and growing pressure from healthcare providers who simply will not work with uncertified suppliers. ISO 13485 certification gives your organisation the documented credibility regulators and customers expect.
As device complexity increases and regulatory pathways become more demanding, certified organisations are better positioned to bring products to market faster, respond to regulatory changes with confidence, and build the kind of trust that healthcare providers and patients are right to expect.
We provide deep, sector-specific auditing aligned directly with your daily operations.
Build the internal expertise your organisation needs to implement, maintain, and audit ISO 13485 effectively.
For professionals looking to conduct and lead ISO 13485 audits with confidence and internationally recognised credentials.
For those responsible for designing, implementing, and maintaining a medical device QMS within their organisation.
For teams and individuals who need a clear, practical understanding of ISO 13485 and what it means for quality and compliance in their day-to-day work.
We are not just a certification body, we are the partner that helps your organisation earn trust, improve performance, and grow with confidence.
Accredited by the ABIS (Accreditation Board for International Standards), our certificates are accepted by clients, regulators, and procurement bodies worldwide.
Our auditors bring sector-specific knowledge to every engagement, ensuring your audit is relevant, thorough, and conducted by someone who understands your business.
We operate globally with the capability to conduct both remote and on-site audits, delivering consistent, high-quality certification services wherever you are.
From your first enquiry to your final certificate, we keep things clear, efficient, and tailored to your organisation - no unnecessary delays, no hidden costs.
ISO 13485 certification is independent confirmation that your Quality Management System meets the requirements of the standard written specifically for medical devices. For most manufacturers, it is not optional in any practical sense, it is the foundation regulators and customers expect to see before a device reaches the market.
No. While the two standards share some structural similarities, ISO 13485 is built specifically around medical device regulatory requirements and risk management, rather than general customer satisfaction and continual improvement. Many organisations hold both, but ISO 13485 cannot simply be treated as an extension of ISO 9001.
Most organisations complete certification within 3 to 6 months, though this can extend depending on device classification, the maturity of your design and risk files, and how many markets you need to address. We will give you a realistic estimate once we understand your scope.
Costs depend on your organisation's size, device classification, number of sites, and process complexity. Pacific Certifications offers transparent, competitive pricing with no hidden fees. Contact us or use our cost calculator for a tailored estimate.
Certification itself is technically voluntary, but in practice, most regulatory pathways and customer relationships in the medical device industry treat it as essential. Without it, getting a device approved or distributed in major markets becomes extremely difficult.
Certification itself is technically voluntary, but in practice, most regulatory pathways and customer relationships in the medical device industry treat it as essential. Without it, getting a device approved or distributed in major markets becomes extremely difficult.
Certificates are valid for three years, with annual surveillance audits conducted throughout that period to confirm your QMS remains effective. A full recertification audit takes place at the end of the three-year cycle.
Some elements can be conducted remotely, but given the nature of medical device production and the regulatory expectations involved, on-site audits are often necessary, particularly for manufacturing and sterilisation facilities. We will advise on the right approach for your specific operation.
Yes. The standard applies regardless of size, from small design houses and startups to large multinational manufacturers. What matters is that your QMS is appropriately scaled to your role in the device lifecycle and genuinely implemented.
To a degree. While ISO 13485 is not built on the Annex SL framework, organisations holding both standards can often align audit scheduling and reduce some duplication, though each standard's specific requirements must still be fully addressed on their own terms.
If non-conformities are identified, you will be given a defined timeframe to investigate, correct, and provide evidence of resolution. Our auditors work with you constructively throughout this process, our goal is to help you reach certification, not to make the path harder than it needs to be.
Get in touch with us today. Complete the form and we’ll be happy to assist you.
This will close in 20 seconds
WhatsApp us

